Claudia Krusche, Sebastian Kant, Hoda Moazzen
with Joana Ahlburhg, Alma de Angelis and Sophia Wüllenweber
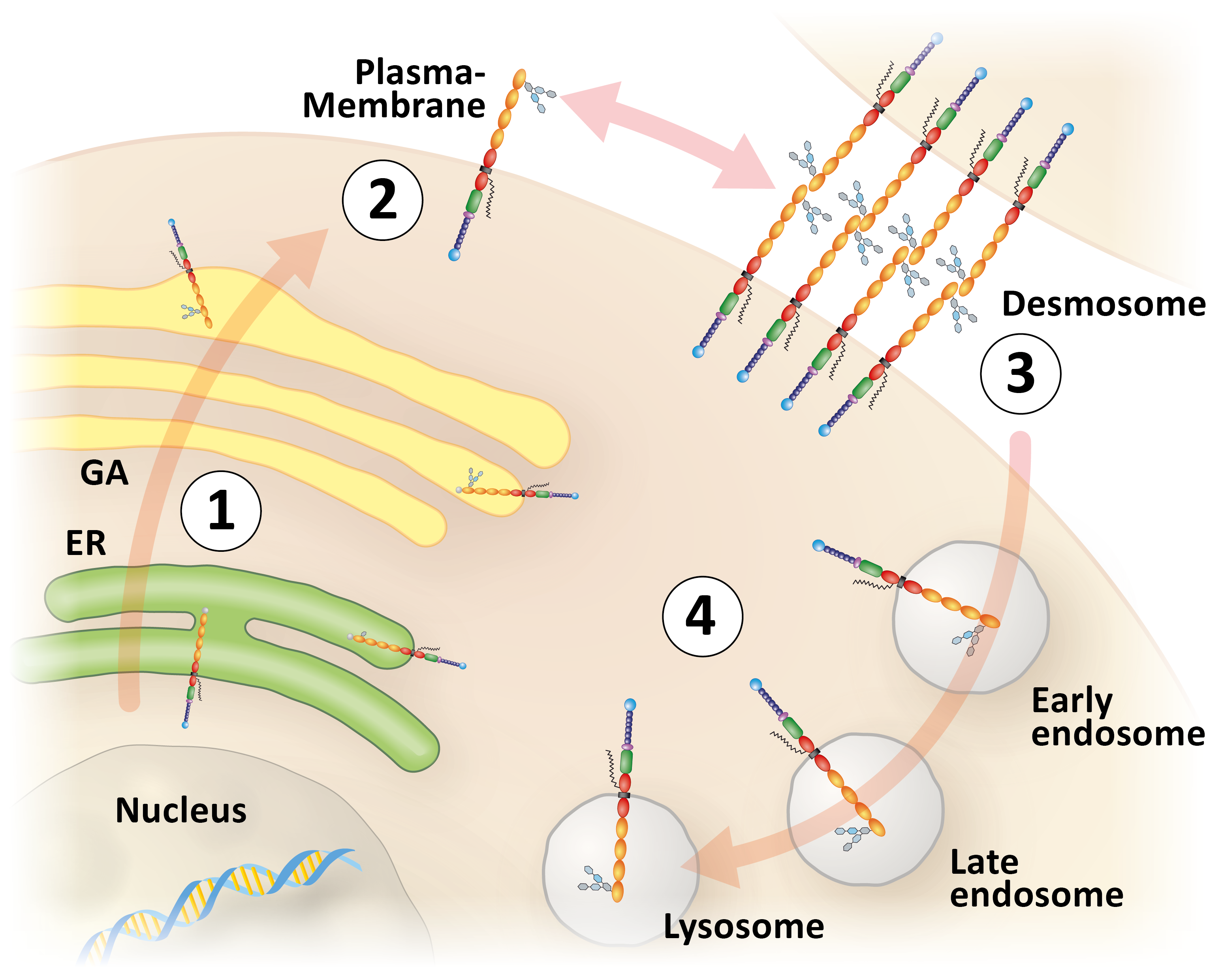
Figure 1. Schematic representation of desmoglein 2-trafficking starting with protein biosynthesis in the endoplasmic reticulum (ER; 1), vesicular transport to the plasma membrane via the Golgi Apparatus (GA; 2), subsequent integration into desmosomes (3), endocytotic uptake into early endosomes, which mature through late endosomes into degradative lysosomes (4).
The group of Sebastian Kant is interested in elucidating the molecular mechanisms determining desmosome formation and their contribution to cell adhesion with a focus on desmosomal cadherins. This is done by using fluorescent reporters to monitor different stages of desmosome assembly and disassembly (Figure 1) and by using cadherin-deficient cultured cells that are rescued with wild-type and mutant desmosomal cadherins many of which have been linked etiologically to human cardiomyopathies. These experiments are complemented by observations in transgenic mice with targeted desmosomal cadherin depletion or mutations, which induce a cardiomyopathy phenotype. The consequences on cell adhesion, contractility, signaling and cellular vitality are examined in the different paradigms.
The passion of Hoda Moazzen's group is to unravel embryonic heart morphogenesis. We are eager to unveil the genetic regulation and intracellular signaling pathways that build a four chambered heart. Annually, 70 infants per 10 000 live births are born with heart disease. This accounts for nearly 36 000 infants annually only in the European Union. Therefore, enhancing our knowledge on regulators of heart development and factors that can induce lifelong heart diseases is a necessity. To address this challenge, we use mice mimicking human heart disease. Specifically, we characterize the cellular and morphological events that occur in embryonic hearts as a result of genetic alterations in desmosomal proteins (example in Figure 2). Furthermore, we aim to transfer the knowledge obtained from embryonic stages to adult heart diseases and to identify approaches for therapeutic treatments.
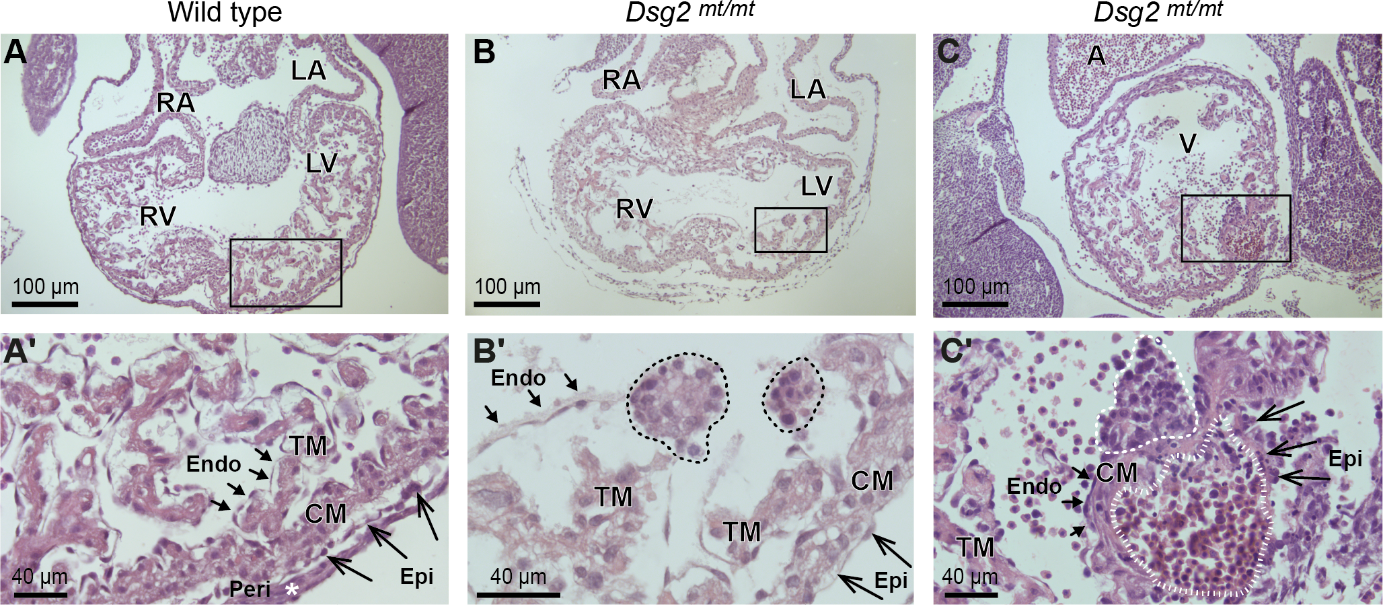
Figure 2. Histological comparison of wild-type and desmoglein 2-mutant (Dsg2 mt/mt) embryonic hearts reveals two types of pathological cell clusters in the mutants. The images show hematoxylin/eosin-stained heart sections at embryonic day E11.5 at low and high magnification. The boxed areas in the upper row correspond to the magnified regions below. Prominent structures, regions and cell layers are labeled: RV, right ventricle; LV, left ventricle; RA, right atrium; LA, left atrium; A, atrium; V, ventricle; Endo, endocardium (short arrows); Epi, epicardium (long arrows); Peri, pericardium; CM, compact myocardium; TM, trabecular myocardium. (A, A') Exemplify wild-type heart morphology. (B-B') The dashed lines delineate two abnormal "type A" cell clusters in Dsg2 mt/mt hearts consisting of densely packed cells that are in continuity with the endocardium. (C-C') depicts a type A cell cluster (dashed line) next to a "type B" cell cluster (striated line) containing numerous nucleated erythrocytes in the subepicardium of a Dsg2 mt/mt heart.
)
Figure 3. Pathology of desmoglein 2-mutant murine hearts. Upper left: Photomicrograph of dissected heart revealing enlarged right ventricle (RV) and prominent fibrotic regions (fibrosis). RA, right atrium; LV, left ventricle; *, regions adjacent to fibrotic foci; remote, region not directly adjacent to fibrotic foci; LA, left atrium. Upper right: Double immunostaining detecting the endothelial cell marker CD31 (brown) and the proliferation antigen Ki67 (pink) as indicators of capillarization and fibroblast proliferation in the fibrotic scar tissue located in the center of the section (counterstain: hematoxylin). Lower left: Electron microscopy showing disrupted cell-cell adhesions (*). Lower right: Tukey’s whisker plots show the results of qRT-PCR (normalized relative quantification; NRQ) performed on RNA isolates from right and left ventricles of Dsg2mt/mt and wild-type controls (dotted lines). Expression was assessed by using non-parametric Mann Whitney tests: *p < 0.05, **p < 0.01; §p > 0.05 and < 0.06. Note the increase of Cd45 (immune cells) and Cd3e (T cells), which is more pronounced in the right ventricle.
Continuous and adaptable heart action is an essential prerequisite for human life. The heart is composed of contractile cardiomyocytes that interact with multiple other cells. A sophisticated cytoskeletal network of desmosome-anchored desmin intermediate filaments plays a fundamental role in cardiomyocyte maturation and maintenance of their highly organized 3D structure supporting lifelong cardiomyocyte contractility. Mutations of desmosomal proteins and their associated desmin intermediate filaments cause arrhythmogenic cardiomyopathy (ACM), which affects 1 in every 2500 young adults. ACM patients initially present with arrhythmia, which leads occasionally to sudden cardiac death. Disease progression is characterized by cardiomyocyte loss, right ventricular dilation, heart wall thinning and ventricular fibrosis resulting in terminal heart failure in severe cases.
The group of Claudia Krusche aims to identify the first mutation-induced pathophysiological events affecting signaling pathways, metabolism, calcium cycling and ultrastructure. In addition, we are studying the molecular processes in disease progression which include chronic inflammation and expansion of interstitial fibrosis. Finally, we study the effects of genetic background and modifier genes on ACM pathophysiology. Our research is motivated by the hope to discover new therapeutical targets to prevent ACM disease onset and to delay its progress.